

Las NACHs son un grupo de trastornos neurológicos hereditarios en los que el hierro se acumula en los ganglios basales, produciendo distonía progresiva, espasticidad, parkinsonismo, anomalías neuropsiquiátricas, atrofia óptica o degeneración de la retina, y a menudo la muerte temprana de los afectados. La forma más prevalente es la neurodegeneración asociada a pantotenato quinasa (PKAN), sobre la que han realizad este estudio expertos de la Universidad Pablo de Olavide. De momento, no existe cura ni un tratamiento estándar para la PKAN.
El equipo científico de la Universidad Pablo de Olavide dirigido por el doctor José Antonio Sánchez Alcázar ha publicado un estudio en la revista internacional Orphanet Journal of Rare Diseases en el que se demuestra que varios compuestos comerciales pueden corregir significativamente el fenotipo mutante en modelos celulares de PKAN. Los suplementos comerciales, pantotenato, pantetina, vitamina E, omega 3, carnitina y tiamina fueron capaces de eliminar la acumulación de hierro y mejorar las alteraciones patológicas en las células mutantes con niveles de expresión residuales del enzima mutado.
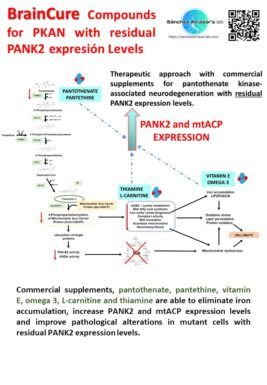
Estos compuestos solos o en combinación son de uso común en la práctica clínica y pueden ser útiles para el tratamiento de pacientes con PKAN con niveles de expresión de enzimas residuales. PKAN es uno de los 15 subtipos de la enfermedad rara denominada neurodegeneración con acumulación de hierro (NACH o NBIA por sus siglas en inglés) que afecta principalmente a niños y niñas en su primera década de vida.
Con el apoyo del Instituto de Salud Carlos III (PI16/00786 y FIS PI19/00377 y Fondo Europeo de Desarrollo Regional, FEDER-Unión Europea), la Junta de Andalucía (CTS-5725 y PY18-850), la Fundación MERCK Salud y FEDER (Federación Española de Enfermedades Raras), este equipo de investigación continúa avanzando en sus estudios sobre las NACHs.
Las NACHs son un grupo de trastornos neurológicos hereditarios en los que el hierro se acumula en los ganglios basales, lo que produce distonía progresiva, espasticidad, parkinsonismo, anomalías neuropsiquiátricas, atrofia óptica o degeneración de la retina, y a menudo la muerte temprana de los afectados. La forma más prevalente de NACH es la neurodegeneración asociada a pantotenato quinasa (PKAN) asociada con mutaciones en el gen de la pantotenato quinasa 2 (PANK2) que es esencial para la síntesis de coenzima A (CoA). No existe cura para la PKAN, ni existe un tratamiento estándar.
Los investigadores de la UPO han demostrado en los fibroblastos y células neurales derivados de los pacientes PKAN que la deficiencia del enzima provoca una acumulación de hierro y una severa deficiencia en la función de diversos procesos mitocondriales esenciales para la producción de energía. Concretamente se ve afectada la síntesis de lípidos mitocondriales, la formación de proteínas con centros hierro-azufre y la actividad de la piruvato deshidrogenasa y del complejo I mitocondrial, todos ellos procesos esenciales en la función mitocondrial.

Equipo de investigación de José Antonio Sánchez Alcázar.
En su trabajo, el equipo ha identificado seis compuestos comerciales capaces de eliminar la acumulación de hierro y aumentar los niveles de expresión del enzima mutante PANK2. El aumento de los niveles de expresión del enzima mutante se asoció con una mejora significativa en las principales alteraciones patológicas de las células PKAN.
La suplementación con pantotenato, pantetina, vitamina E, omega 3, carnitina y/o tiamina puede ser de ayuda para el tratamiento de pacientes con PKAN con niveles de expresión residual de PANK2.
Igualmente es de gran interés que en el trabajo de investigación se ha estudiado la mutación en homocigosis c.680A> G (p.Y227C) con alta prevalencia en determinadas zonas de la Republica Dominicana. Las células portadoras de esta mutación responden favorablemente a los compuestos propuestos.
Las evaluaciones personalizadas en modelos celulares derivados de pacientes pueden ser útiles para evaluar el comportamiento de mutaciones particulares bajo diferentes opciones terapéuticas y, por lo tanto, seleccionar los suplementos y las concentraciones de dosis más efectivos teniendo en cuenta sus propiedades farmacocinéticas.
Proyecto BRAINCURE
Estas investigaciones se enmarcan en el Proyecto BrainCure desarrollado por el equipo científico dirigido por el profesor de la UPO José Antonio Sánchez Alcázar, del Departamento de Fisiología, Anatomía y Biología Celular.
Desde que se pusiera en marcha este proyecto en el año 2014, el equipo del profesor Sánchez Alcázar, que desarrolla su trabajo en el Centro Andaluz de Biología del Desarrollo (CABD), ha logrado grandes avances. Estos científicos y científicas proponen una visión ambiciosa y adaptada al nuevo concepto de medicina personalizada. Así, su trabajo se centra en evaluar la efectividad terapéutica de los distintos tratamientos en los fibroblastos derivados de los pacientes y en las células neuronales generadas por reprogramación directa.
De esta manera, los resultados obtenidos en el laboratorio con modelos celulares de los propios pacientes podrán ser trasladado a la clínica para la realización de ensayos clínicos controlados.
En la actualidad, el Proyecto BrainCure está realizando medicina de precisión en los 5 subtipos más frecuentes: PKAN, neurodegeneración asociada a pantotenato kinasa, con mutaciones en el gen PANK2; PLAN, neurodegeneración asociada a PLA2G6, con mutaciones en el gen PLA2G6; BPAN, neurodegeneración asociada a la proteína beta-propeller, con mutaciones en el gen WDR45; MPAN, neurodegeneración asociada a la proteína de la membrana mitocondrial, con mutaciones en el gen C19orf12; y FAHN, neurodegeneración asociada a la hidroxilasa de ácidos grasos, con mutaciones en el gen FA2H.
El equipo científico está realizando actualmente medicina personalizada en más de 40 pacientes procedentes de España y otros países como Brasil, Colombia, México, EEUU, Francia, Reino Unido, Holanda, Hungría y Polonia.
Más información:
Therapeutic approach with commercial supplements for pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Mónica Álvarez-Córdoba, Diana Reche-López, Paula Cilleros-Holgado, Marta Talaverón-Rey, Irene Villalón-García, Suleva Povea-Cabello, Juan M. Suárez-Rivero, Alejandra Suárez-Carrillo, Manuel Munuera-Cabeza, Rocío Piñero-Pérez, and José A. Sánchez Alcázar. Orphanet Journal of Rare Diseases 2022 Aug 9;17(1):311. doi: 10.1186/s13023-022-02465-9
Referencias del grupo de investigación:
https://pubmed.ncbi.nlm.nih.gov/
https://www.researchgate.net/profile/Jose-Sanchez-Alcazar


Suscríbete a nuestra newsletter
y recibe el mejor contenido de i+Descubre directo a tu email
