
El equipo científico dirigido por el profesor de la Universidad Pablo de Olavide José Antonio Sánchez Alcázar, en colaboración con los profesores, también de la UPO, Antonio Rodríguez Moreno y José Ángel Armengol, han publicado recientemente un estudio que muestra una conversión directa exitosa de fibroblastos derivados de pacientes MELAS en neuronas inducidas.
Las enfermedades mitocondriales son un grupo heterogéneo de trastornos genéticos raros causados por mutaciones en el ADN nuclear o mitocondrial (ADNmt). Estas enfermedades son frecuentemente multisistémicas, aunque afectan principalmente a tejidos que requieren grandes cantidades de energía como el cerebro. Las mutaciones en el ARN de transferencia mitocondrial (mt-tRNA) conducen a defectos en la traducción de proteínas que pueden comprometer algunas o todas las proteínas codificadas por el mtDNA.
El síndrome de encefalomiopatía mitocondrial, acidosis láctica y episodios similares a accidentes cerebrovasculares (MELAS) está causado principalmente por la mutación m.3243A>G en el gen mt-tRNALeu(UUR) (MT-TL1). Actualmente, no existen tratamientos curativos para el síndrome MELAS por lo que es necesario el desarrollo de modelos de la enfermedad que permitan la búsqueda de terapias efectivas.
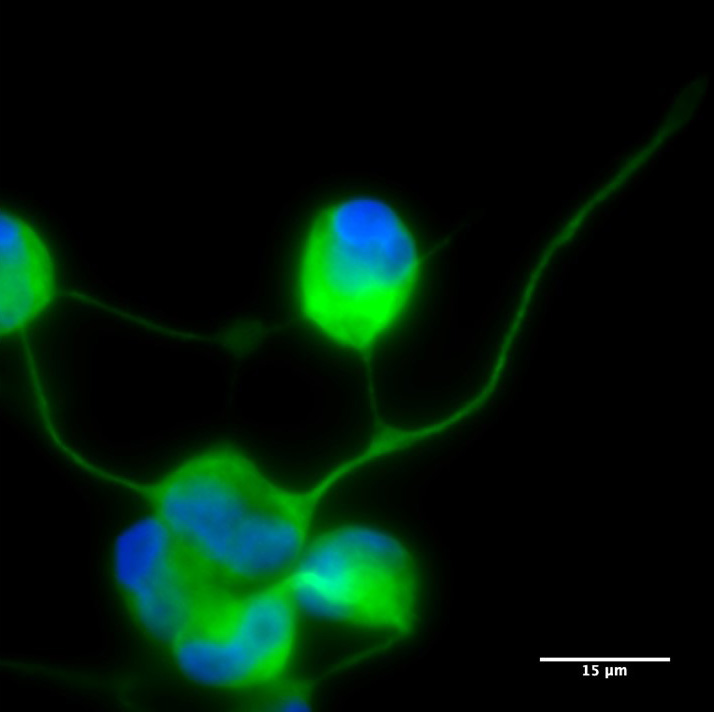
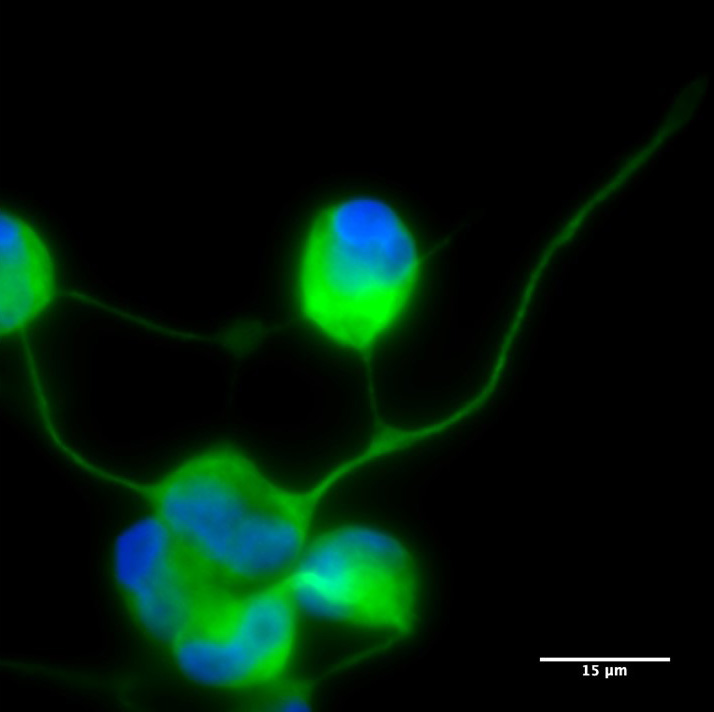
Neurona de paciente MELAS obtenida por reprogramación directa y cocultivada con astrocitos.
Debido a la falta de modelos animales adecuados, se han desarrollado varios modelos celulares para estudiar la enfermedad, proporcionando información sobre los mecanismos fisiopatológicos de MELAS.
En este contexto, el equipo científico dirigido por el profesor de la Universidad Pablo de Olavide José Antonio Sánchez Alcázar, en colaboración con los profesores, también de la UPO, Antonio Rodríguez Moreno y José Ángel Armengol, han publicado recientemente un estudio que muestra una conversión directa exitosa de fibroblastos derivados de pacientes MELAS en neuronas inducidas.
Las neuronas generadas mantienen la mutación presente en las células de la piel fibroblastos y, cocultivados con astrocitos (células de soporte que proporcionan nutrición, apoyo y protección a las neuronas), muestran propiedades electrofisiológicas.
Por lo tanto, la reprogramación directa se confirma como una estrategia prometedora para los estudios de las enfermedades mitocondriales y su utilización para cribado farmacológicos.
De esta forma, el equipo de investigación ha desarrollado un modelo con uno de los tipos celulares más afectados en la enfermedad lo que permitirá una mejor comprensión de los mecanismos patológicos de la enfermedad y la evaluación de potenciales tratamientos.
Plataforma MITOCURE: Medicina de precisión personalizada en las enfermedades mitocondriales
Las enfermedades mitocondriales abarcan un amplio espectro de trastornos musculares y neurodegenerativos, crónicos y progresivos, causadas por mutaciones en el ADN nuclear (nDNA) o mitocondrial (mtDNA), la mayoría de las cuales no tienen tratamiento eficaz. Estas enfermedades tienen una gran heterogeneidad y afectan fundamentalmente a la capacidad de producción energética de las células.
Las terapias farmacológicas actuales se basan fundamentalmente en: 1) Eliminar los metabolitos tóxicos; 2) Intentar circunvalar los bloqueos de la cadena respiratoria; 3) Administrar metabolitos y cofactores para mejorar la síntesis de ATP; 4) Prevenir el estrés oxidativo.
Dada la diversidad de mutaciones y las diferentes opciones terapéuticas, nuestra propuesta defiende que en las enfermedades mitocondriales es obligado una aproximación terapéutica personalizada.
Con esta plataforma los investigadores de la UPO evalúan la efectividad terapéutica de los distintos tratamientos disponibles en los fibroblastos derivados de los pacientes mitocondriales y en las células neuronales generadas por reprogramación directa. Para conseguir este objetivo, estudian los efectos de estos tratamientos sobre las alteraciones fisiopatológicas presentes en los fibroblastos y células neuronales derivadas de los pacientes de una manera personalizada.
En los modelos celulares estudian la proliferación celular y/o muerte celular en medio con galactosa (que fuerza la obtención de energía por la mitocondria), las actividades enzimáticas de la cadena respiratoria mitocondrial, los niveles de expresión de las proteínas mitocondriales, el potencial de membrana mitocondrial, y la activación de la degradación de las mitocondrias y/o la apoptosis.
En la actualidad la plataforma MITOCURE está realizando medición de precisión personalizada en más de 30 mutaciones que afectan directa o indirectamente a la formación de energía por las mitocondrias.
Artículo de referencia:
Povea-Cabello S, Villanueva-Paz M, Villalón-García I, Talaverón-Rey M, Álvarez-Cordoba M, Suárez-Rivero JM, Montes MÁ, Rodríguez-Moreno A, Andrade-Talavera Y, Armengol JA, Sánchez-Alcázar JA. Modeling Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes Syndrome Using Patient-Derived Induced Neurons Generated by Direct Reprogramming. Cell Reprogram. 2022 Jul 8. doi: 10.1089/cell.2022.0055.


Suscríbete a nuestra newsletter
y recibe el mejor contenido de i+Descubre directo a tu email
