
Nuevo modelo experimental para estudiar un tipo de atrofia humana del nervio óptico
Un estudio internacional con participación de investigadores de la Universidad Pablo de Olavide, de Sevilla, ha conseguido generar un ratón mutante que recrea el síndrome de Bosch-Boonstra-Schaaf (BBSOA) y, por ende, permite reproducir la sintomatología básica de la atrofia del nervio óptico y la consiguiente pérdida de la función visual de los pacientes.
Un estudio internacional con participación de investigadores de la Universidad Pablo de Olavide, de Sevilla, ha conseguido generar un ratón mutante que recrea el síndrome de Bosch-Boonstra-Schaaf (BBSOA) y, por ende, permite reproducir la sintomatología básica de la atrofia del nervio óptico y la consiguiente pérdida de la función visual de los pacientes.
Esta enfermedad rara, que se origina por la mutación del gen NR2F1, es un proceso neurodegenerativo que se caracteriza por retraso en el desarrollo, discapacidad intelectual moderada y que afecta el nervio óptico, que es el encargado de transmitir el impulso nervioso desde la retina hacia el cerebro.
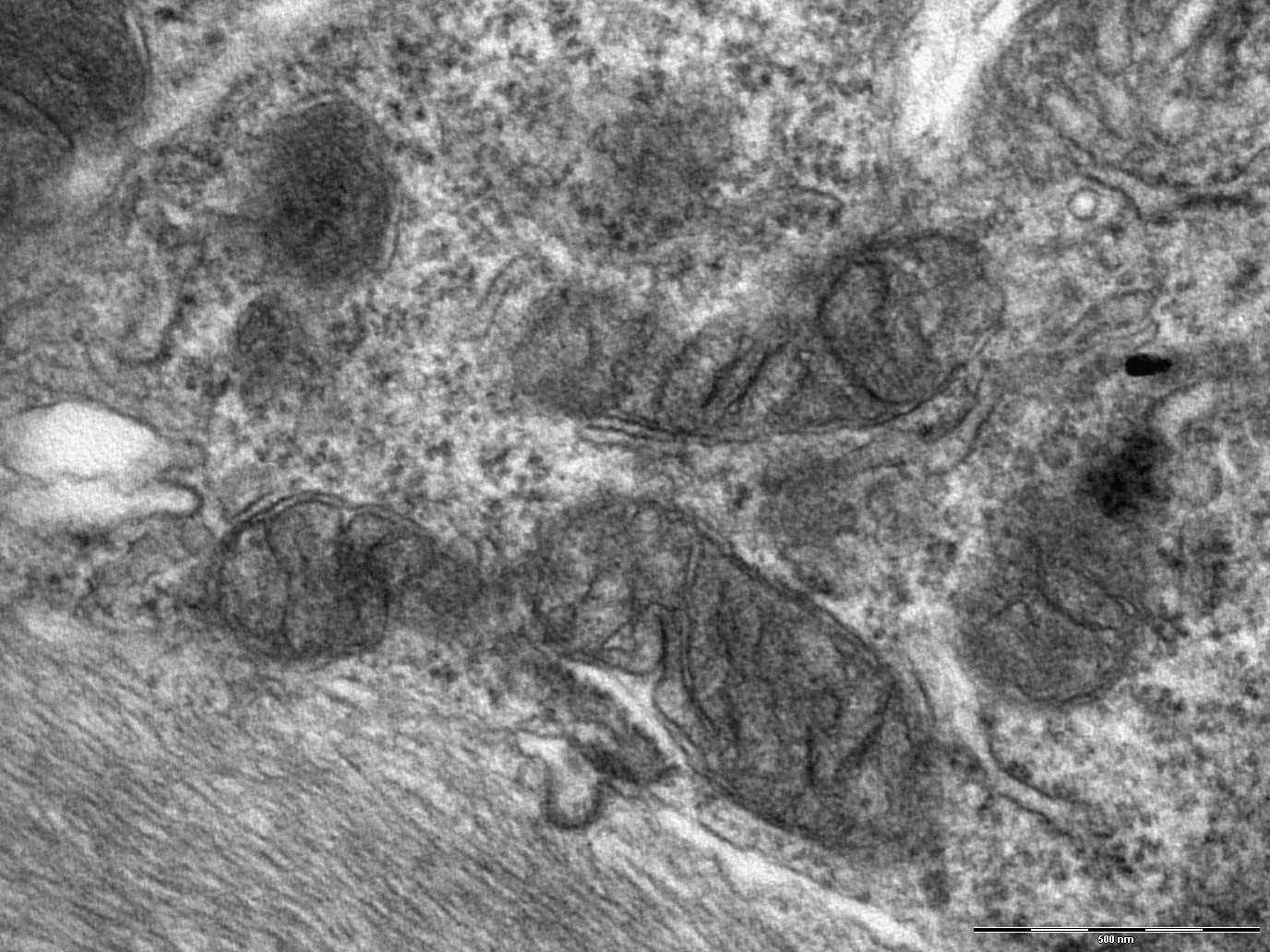
Micrografia electronica de un astrocito hipertrofico en el nervio optico de un raton deficiente en Nr2f1. Foto: P. Bovolenta.
Publicado en la prestigiosa revista EMBO Molecular Medicine, este estudio aporta nuevos conocimientos sobre los mecanismos celulares de la atrofia del nervio óptico en pacientes con BBSOA y abren una vía prometedora para futuros enfoques terapéuticos. Dado su interés, ha recibido un comentario de los editores y le han concedido la portada de la citada revista internacional.
En esta investigación, coordinada por el equipo del Instituto Nacional de Salud e Investigación Médica (Inserm) francés que lidera Michèle Studer en el Instituto de Biología de Valrose, han participado los científicos de la División de Neurociencias de la Universidad Pablo de Olavide José María Delgado y Agnès Gruart, el grupo de investigación liderado por Paola Bovolenta en el Centro de Biología Molecular Severo Ochoa (centro mixto del CSIC y la Universidad Autónoma de Madrid) y el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER).
El principal objetivo del estudio fue recrear en el laboratorio un modelo de esta enfermedad a fin de poder diseccionar los mecanismos celulares y moleculares que conducen a una disminución gradual de la agudeza visual en los niños afectados. Los grupos de la Universidad de Niza y del Consejo Superior de Investigaciones Científicas estudiaron en detalle el desarrollo del nervio óptico en el ratón normal y tras la mutación del gen NR2F1.
El nervio óptico conecta la retina con el resto de las estructuras cerebrales importantes para la visión, por lo que es fundamental para un perfecto funcionamiento del sistema visual en el adulto. Ambos grupos demostraron degeneración o atrofia del nervio óptico en los ratones mientras que el grupo de investigadores de la Universidad Pablo de Olavide (UPO) demostró que los ratones mutantes adultos presentan un retraso en la conducción de los impulsos nerviosos por la vía visual y evidentes limitaciones funcionales en pruebas de aprendizaje que requieren una percepción visual normal. Toda esta sintomatología reproduce con fidelidad el síndrome de la atrofia del nervio óptico en pacientes.
La investigación sugiere que algunas de las características clínicas de esta enfermedad podrían abordarse con un tratamiento con medicamentos químicos en la vida postnatal temprana. “Como, al parecer, estos déficits funcionales se deben a una disfunción de las células -denominadas oligodendrocitos- que envuelven al nervio óptico, en nuestro estudio se ha mostrado que la administración de Miconazol en ratones mutantes mejora la disponibilidad de estas células y, por tanto, la función visual. Queda por comprobar si este tratamiento podrá validarse en un futuro próximo como efectivo en humanos”, afirma el investigador de la UPO José María Delgado.


Suscríbete a nuestra newsletter
y recibe el mejor contenido de i+Descubre directo a tu email
